Comparative Study of Monolayer Fatty Acids at the Air/Water Interface
Bianca Monica RUS
Technical University of Cluj-Napoca, Romania
Abstract
The behavior of insoluble monolayer of surfactants spread at the air/water interfaces is frequently studied by recording compression isotherms. The compression isotherms given in terms of surface pressure versus mean molecular area are well reproducible for a chosen spreading molecular area of fatty acids, like stearic acid, oleic acid and linoleic acid.
Mathematical modeling of these isotherms is performed by means of surface state equations. In the literature, a great variety of such equations have been proposed. In the present paper is made a study of the compression isotherms of oleic acid (OA) monolayer spread at the air /water interface on acidic aqueous solutions (pH = 2).
Keywords
Fatty Acid Monolayer, Equations of State, Air/water Interface
I. Introduction
The thermodynamic and structural characterization of fatty acid films at the air-water interface has received a remarkable attention over the past several decades [1-6]. The thermodynamic analysis of compression isotherms in terms of surface pressure (π, mN/m) versus mean molecular area (A, nm2/molecule) leads to equations of state that describes well the phase behavior of fatty acid monolayer.
At very low surface pressures, monolayer might be considered to behave like two-dimensional gases. Therefore, in these conditions, the validity of the two-dimensional perfect gas state equation [4]:
πA = kT (1)
may be expected, where k is Boltzmanns constant and T is the absolute temperature. Due to the experimental difficulties in measuring very low surface pressures, this region is less investigated.
Much more data are available in the range of 0.2 mN/m < π <10 mN/m, where monolayer generally are in a liquid expanded state. Since the cross-sectional area of the surfactant molecules cannot be neglected and it represents a mean area necessity named also co-area, in equation (1) the A value has been corrected for a co-area (A0), obtaining [9]:
π(A - A0) = kT (2)
where A0 is frequently taken as an adjusted parameter.
Since in the liquid state the intermolecular interactions might be more important than the own molecular area of the surfactant, an internal pressure, π0, can be introduced. Correcting π in equation (1) for this internal pressure, one obtains:
(π+π0)A = kT (3)
By taking into account both intermolecular attractive forces and the above mentioned co-area, the state equation (1) becomes [10]:
(π +π0)(A - A0) = kT (4)
Further, a two-dimensional state equation, analogous with the Van der Waals equation:
(π + α/A2)(A - A0) = kT (5)
has also been proposed. Equation (5) results from equation (4), by taking π0 = α/A2 on the base of the same reason, which in the case of a bulk gaseous phase leads to the presumption that the three-dimensional internal pressure is inversely proportional with the square of the volume.
There were made several attempts to correlate the parameter α with molecular structure and with intermolecular interactions. On the basis of the scaled particle theory for fatty acids the state equation:
(π +α/A2)[A (1 A0'/A)2] = kT (6)
was derived [11]. In this equation A0' = πd2/4 represents the area of a hard disc having its diameter equal to d, which is thought to be equivalent with the cross-sectional area of the saturated hydrocarbon chain. The correlation between A0' and the co-area A0 is A0'=A0/2. The parameter α was also correlated with molecular and intermolecular characteristics.
For cohering uncharged films the equation:
(π + α/A3/2) (A - A0) = kT (7)
was derived [12] and it was found to be in good agreement with some experimental results.
According to the duplex film model, a surfactant monolayer spread at the air/water interface may be considered as a monolayer solution formed of the surfactant head groups dissolved in water and having the theoretical surface pressure πh, covered by an oily layer consisting of the hydrophobic chains of surfactant molecules. Denoting by (-π0) the spreading coefficient of this oil on the hypothetical surface solution of surfactant head groups and interfacial water, the overall surface pressure may be expressed as:
π = -π0 + πh (8)
By considering an insoluble monolayer, on the basis of the equality of the chemical potential of water in the bulk subphase and in the monolayer solution of headgroups, πh may be given by a Butler type equation [13]:
![]() (9)
(9)
where, A1, a1, γ1 and x1 stands for the molecular area, activity, activity coefficient and molar fraction of water molecules in the monolayer solution, respectively. Combination of equation (8) and (9) yields [14]:
![]() (10)
(10)
By considering the headgroup monolayer to be a binary regular solution, one has
![]() (11)
(11)
where β12 and x2 stands for the interaction parameter between the polar head group and water molecules in the monolayer and for the molar fraction of the head groups in the monolayer, respectively.
Finally, the following equation of state is obtained:
![]() (12)
(12)
It is well to mention that other state equations have also been derived based on statistical considerations of the conformation of hydrocarbon chain segments [15, 16].
II. Material and Methods
In view of testing the state equations the OA monolayer has been chosen as model system. The compression isotherm recorded on a subphase with pH = 2 has been used since at this pH the protolytic equilibrium in the monolayer is shifted practically completely towards the neutral OA molecules [7]. The experimental π vs. A curve is given in fig.1 (curve 1). The theoretical curve, corresponding to equation (1) is visualized in the same figure (curve 2). As seen, at high A values important deviations from the perfect behavior appear in the negative direction, indicating the role of the intermolecular attractive forces. On the contrary, at lower A values, the deviations become positive.
II. Results and Discussion
The other state equations contain single (equations (2) and (3) or even two equations 4-7) parameters to be derived from experimental data. Since a good mathematical description of the compression isotherms by means of state equations can be expected in the case of expanded films, i.e. at low surface pressures, the π ≤ 7 mN/m has been chosen. This domain is delimited in Fig. 1 by dashed straight lines, and we had in this region a number of 17 experimental points, i.e. Ai - πi pairs. These data were processed following a curve fitting method. For this purpose, the standard deviation □ of the experimental points from the theoretical ones was calculated and minimized by performing a systematic variation of the parameter / parameters to be derived.
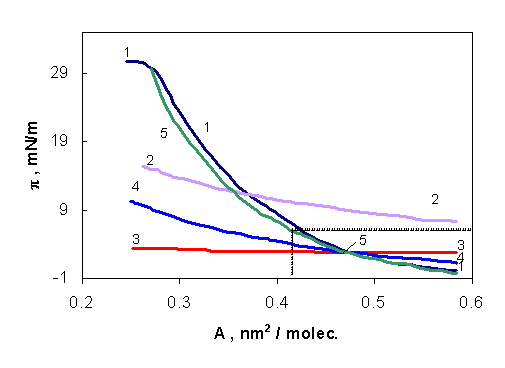
Fig. 1. πvs. A isotherms for OA monolayer. Experimental curve(1);
theoretical curves (2-5) calculated by using parameters given in Table 1 and state equations:
curve (2) equation (1); (3) - equation (2); (4) - equations (3); (5) - equation (4).
In the case of equation (2) and (3), by varying A0 and π0, respectively, the minimum standard deviation, m, was determined, which is given in Table 1, together with the corresponding parameter value. By using the latters the theoretical ones, given in Fig. 1, were calculated (curves 3 and 4). As seen, equation (2) gives a rather unrealistic, negative A0 value, while equation (3) yields a π0 value corresponding to intermolecular attraction. Neither equation (2), nor equation (3) is able to give a good description of the experimental curve, the m value being rather high.
By using equations with two adjustable parameters, a double minimization of □ is to be performed. To this end, one of the parameters is maintained at a constant value, and by varying the other one, the minimum of □, denoted as □m is determined. The systematic variation of the first parameter allows us to determine the minimum of the □m values, (□m)m, indicating the best values of both parameters to be derived. These double minimum values of ∆ are given in Table 1, and the theoretical curve calculated by means of equation (4), using π0 and A0 values given in Table 1, is also visualized in Fig. 1, curve (5). Obviously, the use of two adjustable parameters entails a spectacular improvement of the approximation.
Table 1. Parameters of the state equation and standard deviations
derived for OA monolayer (π ≤ 7 mN/m)
|
Eq |
π0 (mN/m) |
A0 (nm2/molec) |
α* |
β12∙1020 (Nm) |
□** (mN/M) |
|
(1) |
- |
- |
- |
- |
6.09 |
|
(2) |
- |
-1.123 |
- |
- |
1.95 |
|
(3) |
6.0 |
- |
- |
- |
1.19 |
|
(4) |
10.5 |
0.169 |
- |
- |
0.29 |
|
(6) |
- |
0.268 |
5.691 |
- |
0.17 |
|
(7) |
10.46 |
- |
- |
-0.08 |
0.29 |
|
(9) |
- |
- |
0.842 |
-1.07 |
0.15 |
|
(10) |
- |
- |
8.601 |
-2.28 |
0.09 |
|
(11) |
- |
- |
1.153 |
-5.73 |
0.13 |
*Units: 1) 10-30 Nm2; 2) 10 -20 Nm; 3) 10-38 Nm2; in all cases per molecule
**It means m in the case of equations (2) and (3), and (m)m with equations (4), (6), (7) and (9-11).
As seen from Table 1, equation (6) gives better results than the others. As far as equation (7) is concerned, its use raises several problems, related to calculation of the molar fractions in the monolayer.
References
1. Dynarowicz-Latka P., Dhanabalan A., Oliveira O. N., Colloid and Interface Sci., 91 (2), 2001, p. 221-293.
2. Harkins W. D., The Physical Chemistry of Surface Films, Reinhold, New York, 1952.
3. Davies J.T., and Rideal, E.K., Interfacial Phenomena, Academic Press, New York, 1963.
4. Gaines G.L.Jr., Insoluble Monolayers at Liquid-Gas Interface, Wiley-InterScience, New York, 1966.
5. Hiemenz P.C., Principles of Colloid and Surface Chemistry, Dekker, New York, 1966.
6. Lim Y.C., and Berg J.C., J. Colloid and Interface Sci., 51 (1), 1975, p. 162.
7. Tomoaia Cotisel M., Zsako J., Mocanu A., Lupea M., Chifu E., J. Colloid Interface Sci., 117, 1987, p. 464.
8. Tomoaia-Cotisel M., Mocanu A., Pop V.D, Plesa D., Plesa S., Albu I., Equations of State for Films of Fatty Acids at the Air/Water Interface, The VIIth Symposium of Colloid and Surface Chemistry, Bucharest, 2002.
9. Volmer M., Z. Physik. Chem., 115, 1925, p. 253.
10. Langmuir I., J. Chem. Phys., 1 , 1933, p. 756.
11. Reiss H., Frisch H.L., Lebowitz J.L., J. Chem. Phys., 31, 1959, p. 369.
12. Guastalla J., Cahiers Phys., 10, 1942, p. 30 ; J. Chem. Phys., 43, 1946, p. 184.
13. Butler J.A.V., Proc. Royal. Soc. London, Ser. A, 135, 1932, p. 348.
14. Lim Y.C., Berg J.C., J. Colloid Interface Sci., 51, 1975, p. 162.
15. Motomura K., Motomura R., J. Colloid Interface Sci., 29, 1969, p. 617.
16. MacDowell L.G., Vega C., Sanz E., J. Chem. Phys., 115 (13), 2001, p. 6220.