Engineering, Environment
Computational modelling of the kinetics and thermodynamics of Diels-Alder reaction: 1, 3-cyclohexadiene and substituted ethene
Omolara Olubunmi ADEBOYE *1, Isaiah Ajibade ADEJORO 2 and Abimbola Modupe OLATUNDE 2
1 Department of Chemistry, Emmanuel Alayande College of Education, Oyo, Nigeria
2 Department of Chemistry, University of Ibadan, Ibadan, Nigeria
E-mails: moadeb5848@yahoo.com, ajibadejoro@yahoo.com, abimbolatunde1@yahoo.com
*Corresponding Author phone: +2348058484417
Received: November 29, 2017 / Accepted: November 26, 2018 / Published: December 30, 2018
Abstract
The kinetics and thermodynamics of the Diels-Alder reaction of 1-3, Cyclohexadiene and substituted ethene were modelled in the using Density Functional Theory (DFT) at B3LYP level with 6-311++G (2df, 2P) basis set. Electron withdrawing and electron donating group were substituted for hydrogen in ethene (dienophile). Geometric, kinetics, thermodynamic parameters and the HOMO/LUMO energy gap were calculated. The values obtained for the change in the bond length (∆d) in the transition state and the reactants showed that the reaction is asynchronous and energy of formation obtained confirms the exothermicity of the reaction using Hammonds postulate. The result obtained showed that that electron withdrawing group (NO2-substituted dienophile) lowers the activation parameters ΔH*(89.744 kJ/mol); Ea (92.22 kJ/mol); ΔG*( 93.916 kJ/mol) and thereby increasing the rate of the reaction as observed with lower rate constant, k value (2.17 x 10-2 mol-1l t-1) compared with unsubstituted and OH-substituted dienophile(ΔH*109.794,121.768 kJ/mol) Ea (112.273, 124.247 kJ/mol) and ΔG*(115.158, 128.324 kJ/mol) respectively. The Rho (ρ) value calculated to describe the sensitivity of the reactions to substituent effect for OH (-1.777) showed that the reaction is creating a positive charge and for NO2 (+0.223eV) is an indication that the negative charge is building up for the stabilization effect of the reactions. EWG, NO2 results into the decrease of HOMO-LUMO energy gap and this causes an increase in the feasibility of the reaction, it is observed that NO2 with energy gap (-2.91eV) are energetically feasible than the OH substituted dienophile and more than the simple Diels-Alder reaction.
Keywords
Diels Alder reaction; Gas-phase; Substituted ethene
Introduction
Diels-Alder reaction (Figure 1) is a [4+2] cycloaddition reaction which involves a molecule with a conjugated π-system (a diene) and another with at least a π-bond (a dienophile) [1]. Diels Alder reactions are important organic reactions and are extensively used in carbon-carbon bond formation in organic reactions [1-7]. Synthetically, the Diels Alder reactions are important cycloaddition reaction and the most important pericyclic reactions. The study of Diels-Alder reaction has attracted interest [2] because of its importance in diene synthesis and a powerful tool in the synthesis of new organic compounds through a six-membered cyclic system [7-13]. Experimental analysis and theoretical calculations showed that the reactions occur through a concerted mechanism, although some occurs in steps [3, 4, 5].
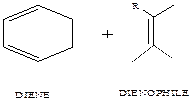
Figure 1. Simple Diels-Alder reaction
The study on prototypical Diels-Alder reactions by hybrid density functional/Hartree-Fock approach showed that the kinetics and the thermodynamic parameters obtained at the B3LYP and QCIST[T] levels approach the experimental value whereas HF and MP2 approaches are not sufficiently reliable [4]. In recent work, substituent effects on simple Diels-Alder reaction as evidence for possible explosive reactions showed that the nature of substituent influences the feasibility of reactions and it also showed the pathway of certain normal and explosive Diels-Alder reactions [6]. Following the work that has been carried out on kinetics and thermodynamics of Diels-Alder reaction there is dearth of information on the influence of substituent attached to the dienophile on the kinetics and thermodynamic parameters.
Therefore, the aim of this work is to use computational modelling using Spartan calculation model with Density Functional Method at B3LYP level using 6-31G* basis set to study the influence of electron withdrawing group and electron donating groups attached to the dienophile on the kinetic and thermodynamic parameters to show the influence on activation parameters.
Materials and method
Computational methodology
Using Spartan 10 density functional method (DFT) with B3LYP/6-31G* basis set, quantum mechanical calculations were carried out on the reaction using the following systematic and consistent computational procedures:
Ø ![]() Conformational search
(MMFF)
Conformational search
(MMFF)
Ø
![]() Reaction path calculation
Reaction path calculation
Ø
![]() Location of the
transition state
Location of the
transition state
Ø
![]() Characterizing the
transition state
Characterizing the
transition state
Ø
![]() Geometric Optimization
Geometric Optimization
Ø Calculations
Conformational search was done on the reactants using molecular mechanics force field (MMFF) to obtain the reactants that has the lowest energy which is indicates the stability of the reactants. Reaction path calculations were carried out using C5-C18 and C2 -C15 as the reaction coordinate this was done by slowly altering the bond distance between C5-C18 and C2 -C15 (5.643Å) to the normal (1.546Å) bond length in the product form in 20 iterations. This was done to obtain the mechanism of the reaction. Optimization was performed on the transition state structure to obtain the true transition state; this was subjected to tests to verify that the transition state structure has only one imaginary vibrational frequency (IR value) and that the saddle point connects the ground state, transition state and product together. The intrinsic reaction coordinates method was also used by optimizing the molecule subject to a fixed position along the reaction coordinate [7]. Full geometry optimization was then carried out on the structure of the reactants, the transition states and products to obtain the geometric parameters such as bond lengths, bond angles, dihedrals and atomic charges.
Kinetics and thermodynamic calculations was done to obtain the kinetic and thermodynamic parameters. To arrive at a closer approximation of the true energy of the molecules, the sum of the ground state energy and the statistically mechanically calculated data was used Eq. [1], [8].
![]() sm (1)
sm (1)
Where: sm - is the statistically mechanically calculated enthalpy, H- the enthalpy of the specie and GSE- ground state energy.
Substitute this into the initial definition of the heat of reaction we have Eq. [2]:
![]() (2)
(2)
Where: ∆Hrxn - change in enthalpy of reaction; GSE - ground state energy of reactants and products and Hsm - statistically mechanically calculated enthalpy of reactants and products.
Activation energy (Ea) was calculated Eq. [3]:
![]() (3)
(3)
Where: Ea - activation energy, ∆H - change in enthalpy, R - gas constant and T temperature.
The entropy of the reaction was calculated by taking the difference of product and reactant entropies that is Eq. [4]:
![]() (4)
(4)
Where: S - entropy and ∆S - change in entropy.
The Gibbs free energy of activation was calculated using the modified version of the heat of reaction equation. Knowing that Eq. [5].
![]() (5)
(5)
Coefficient k was calculated using Eq. [6]:
k
(T) = ![]() (6)
(6)
Where: k' - the Boltzmann; h - Plancks constants.
Energy of HOMO and LUMO was obtained and the band gap was calculated using Eq. [7]:
Band Gap = E HOMO E LUMO (7)
Hammonds postulate was used to explain the exothermicity of the reaction and Rho (ρ) value was calculated to describe the sensitivity of the reaction to substituent using Eq. [8]:
log![]() = ρ
= ρ![]() (8)
(8)
Where: KH - the equilibrium constant for Hydrogen; KX - the equilibrium constant for substituent.
Results and Discussion
Reaction mechanism
The reaction proceeds by
the normal electron demand through a concerted six-membered cyclic transition
state which involves the transfer of electron (breaking of bonds) from three
different ![]() -bonds ( C3=
C5, C2= C7 and C15= C18)
and the formation of one new
-bonds ( C3=
C5, C2= C7 and C15= C18)
and the formation of one new ![]() -bond
between C3 C7 and two new
-bond
between C3 C7 and two new ![]() -bonds
between C2 C15 and C5 C18
(Figure 2).
-bonds
between C2 C15 and C5 C18
(Figure 2).
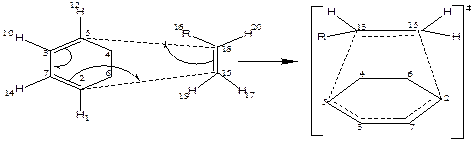
1,3 cyclohexadiene ethene
Figure 2. The reaction mechanism of Diels-Alder reaction of 1,3-cyclohexadiene and substituted ethene
Geometry optimization
Geometry optimization (Figure 3) was done on the reactants, the transition states and the products to obtain the geometric parameters such as bond length, bond angle, dihedral angles and atomic charge distribution (Table 1).
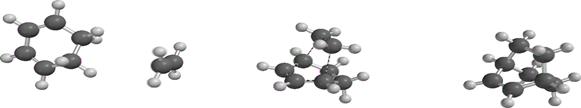
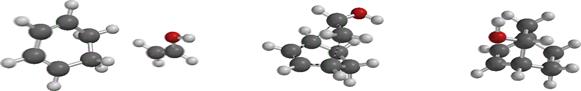
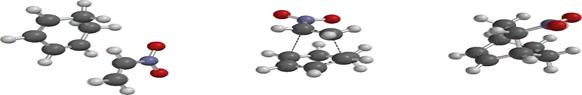
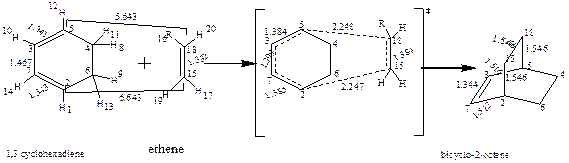
Figure 3. Optimized geometry of the unsubstituted, OH-substituted and NO2- substituted reactants, transition state and product
Figure 3 shows the optimized geometry of the unsubstituted, OH-substituted and NO2-substituted structures of ethane in the reactant, transition and product state to explain the process of bond breaking and bond formation of the reaction between 1,3 cyclohexadiene and ethane to form bicyclo-2-octene. The bond length (Figure 3) is the distance between two atoms. Table 1 shows the bond length value for the reactants, transition state and products respectively. Considering the bond distance for (hydrogen) C3-C5 (1.341, 1.384 and 1.516Å), the values showed that the ground state reflects the value for a carbon-carbon double bond which increased in the transition state with a sharp increase in the value for the product showing the full assumption of a carbon-carbon single bond. In C3-C7 a carbon-carbon single bond exists in the ground state (1.467, 1.399 and 1.344Å) but in the product form a fully formed new pi (π) carbon-carbon double bond emerged. The values obtained in C2-C7 and C15-C18 (1.323, 1.385 and 1.512; 1.332, 1.384 and 1.548 Å respectively) also showed the disappearance of the double bond for the emergence of the carbon-carbon single bonds in the product state. Interestingly, at the ground state, no bond exist between C2 -C15 and C5 and C18, the distances are 6.643 and 5.643 Å but in the transition state it is observed that new bonds are about emerge (2.247 and 2.248 Å) and finally the bond length in the product shows the formation of new carbon-carbon sigma (σ) bonds.
 |
Table 1. Bond length (Å)
|
Bond Length |
States |
H |
OH |
NO2 |
|
C3 C5 |
GS TS PRD ∆d |
1.341 1.384 1.516 +0.043 |
1.388 1.381 1.507 +0.043 |
1.338 1.373 1.509 +0.035 |
|
C3 C7 |
GS TS PRD ∆d |
1.467 1.399 1.344 -0.068 |
1.463 1.396 1.332 -0.087 |
1.464 1.400 1.337 -0.064 |
|
C7 C2 |
GS TS PRD ∆d |
1.343 1.385 1.516 +0.042 |
1.337 1.399 1.508 +0.062 |
1.337 1.397 1.511 +0.060 |
|
C2 H1 |
GS TS PRD ∆d |
1.088 1.083 1.094 -0.005 |
1.083 1.085 1.089 +0.002 |
1.083 1.084 1.088 +0.001 |
|
C5 H12 |
GS TS PRD ∆d |
1.088 1.084 1.094 -0.004 |
1.083 1.084 1.089 +0.001 |
1.083 1.085 1.089 +0.002 |
|
C15 C18 |
GS TS PRD ∆d |
1.332 1.384 1.548 +0.052 |
1.325 1.394 1.549 +0.069 |
1.319 1.393 1.537 +0.074 |
|
C2 C15 |
GS TS PRD ∆d |
5.643 2.247 1.546 -3.396 |
5.851 2.435 1.551 -3.416 |
4.611 2.012 1.556 -2.599 |
|
C5 C18
|
GS TS PRD ∆d |
5.643 2.248 1.546 -3.395 |
5.194 2.066 1.543 -3.484 |
4.793 2.642 1.549 -1.637 |
The bold angle (Table 2) shows the interaction between three atoms to determine the specific arrangement of atoms to explain the stability of the molecule. Variation in the values for the reactants, transition and product states is as a result of the distortion that occurs during transition from reactants through the transition state to the products.
Table 2. Bond angle
|
Bond angle |
States |
H |
OH |
NO2 |
|
H12 - C5 - C3
|
GS TS PRD |
120.69 119.12 112.14 |
120.65 119.57 112.28 |
120.60 119.66 112.36 |
|
C5 - C3 - C7
|
GS TS PRD |
120.64 118.33 114.39 |
120.65 120.71 119.12 |
119.20 114.33 114.38 |
|
H10 - C3 - C7
|
GS TS PRD |
118.71 119.77 123.82 |
118.76 119.82 123.89 |
118.68 119.73 123.74 |
|
C7 - C2 - HI
|
GS TS PRD |
120.69 118.96 112.15 |
120.65 117.87 112.67 |
120.69 117.37 117.56 |
|
C2 - C6 - C4
|
GS TS PRD |
120.33 112.57 109.49 |
120.21 112.36 109.49 |
120.20 112.98 109.29 |
|
C6 - C4 - C5
|
GS TS PRD |
111.72 112.46 109.24 |
111.78 112.12 109.10 |
112.53 112.98 109.29 |
|
C18 - C15 - H17
|
GS TS PRD |
121.77 120.04 110.87 |
121.71 122.62 110.88 |
120.97 116.30 109.58 |
|
H16 -C18-H20 |
GS TS PRD |
116.44 114.98 106.48 |
115.63 111.13 108.76 |
111.91 111.99 102.62 |
The dihedral angle (Table 3) show the interaction between four atoms and also further explains the stability of the structures. The variation in the values for the ground state, transition state and products are also as a result of the distortion that occurs during the transformation from the reactants to the products.
![]() Table 3. Dihedral
angles
Table 3. Dihedral
angles
|
Dihedral angles |
States |
H |
OH |
NO2 |
|
H12 - C5 - C3 - H10 |
GS TS |
-0.88 -6.07 |
-0.90 -1.95 |
0.09 1.67 |
|
C5 - C3 - C7 - C12 |
GS TS |
14.71 1.03 |
14.58 5.48 |
-13.81 5.04 |
|
C6 -C4 -C5 -H12 |
GS TS |
153.21 170.88 |
153.46 170.54 |
-154.21 169.45 |
|
C2 -C6 -C4 -C5 |
GS TS |
43.09 3.85 |
42.84 -2.72 |
-41.06 -6.87 |
|
C15 -C18 -H16 -H20 |
GS TS |
179.92 154.61 |
-178.82 3.34 |
-179.48 -165.04 |
|
C18 -C15 -H17 -H19 |
GS TS |
-179.92 -155.23 |
179.86 -141.66 |
-179.91 140.93 |
The negative values obtained for the enthalpy of reaction (Table 4) ΔH (-111.904, -94.454, and 11.094 for H, OH (electron donating group) and NO2, electron withdrawing group) respectively showed that the reactions are exothermic.
Negative Gibb's free
energy values, ΔG (-99.390, -109.531 and -100.664) showed that the
reactions are spontaneous [12]. The activation parameters ΔH* (109.794,
121.768 and 89.744 kJ/mol); Ea (112.273, 124.247 and 92.22 kJ/mol);
ΔG* (115.158, 128.324 and 93.916 kJ/mol) for H, OH and NO2 as
reported [13] showed that electron withdrawing group on dienophiles lowers the
activation parameters thereby increasing the rate of the reaction as observed
with lower rate constant, k value (2.17 x 10-2) for dienophile
substituted with electron withdrawing group NO2 compared with
dienophile substituted electron donating group. The Rho (![]() )
calculated (OH -1.777) showed the reaction is creating a positive charge and
for NO2 (+0.223) is an indication that the negative charge is
building up for the stabilization effect of the reactions [12].
)
calculated (OH -1.777) showed the reaction is creating a positive charge and
for NO2 (+0.223) is an indication that the negative charge is
building up for the stabilization effect of the reactions [12].
Table 4. Kinetic and thermodynamic parameters
|
Parameters |
H |
OH (EDG) |
NO2 (EWG) |
|
ΔHreaction |
-111.906 |
-94.454 |
-111.094 |
|
ΔSreaction |
-42.299 |
-40.108 |
-26.042 |
|
ΔGreaction |
-99.39 |
-109.53 |
-100.664 |
|
ΔS* |
-17.971 |
-21.289 |
-13.407 |
|
ΔH* |
-109.794 |
-121.768 |
89.744 |
|
Ea |
112.273 |
124.247 |
92.222 |
|
ΔG* |
115.158 |
128.324 |
93.916 |
|
logA |
13.731 |
13.905 |
13.490 |
|
K |
1.156 x 10-6 |
1.3 x 10-7 |
2.17 x 10-2 |
|
Keq |
2.581 x 1017 |
1.545 x 1019 |
4.13 x 1017 |
|
Rho (ρ) |
- |
-1.777 |
+0.223 |
Frontier molecular orbital theory (FMO)
The concertedness and the effect of substituent can be rationalized using FMO. Applying this to this reaction, two modes of interactions are possible, the reaction can be controlled by the interaction of the HOMO of the diene (1, 3-cylohexadiene) with the LUMO of the dienophile (ethene) (the normal electron demand) or by the interaction between the LUMO of the diene and the HOMO of the dienophile (inverse electron demand).
In the former case, a reduction of the diene-HOMO and dienophile-LUMO energy gap (Table 5) can be achieved by raising the energy of the HOMO of the 1,3-cyclohexadiene.
In this reaction, the energy of HOMO (-5.96 eV) is lower compared with the energy of the LUMO (-0.29, -0.49 and -3.06) for the H, OH and NO2 and this may be as a result of the inverse electron demand.
Table 5. HOMO/LUMO Energy gap
|
Elements |
HOMO (Diene) |
LUMO (Dienophile) |
Energy gap |
|
H |
-5.97 |
-0.29 |
-5.68 |
|
OH |
-5.97 |
-0.49 |
-5.48 |
|
NO2 |
-5.97 |
-3.06 |
-2.91 |
In NO2-substituted dienophile, the energy gap is far greater than that of OH substituted dienophile and greater compared with simple Diels Alder reactions. This is in line with result obtained by [13]. EWG, as reported [13] results into the decrease of HOMO-LUMO energy gap and this causes an increase in the feasibility of the reaction, notably with NO2. In this work it is observed that NO2 with energy gap (-2.91) are energetically feasible than the OH substituted dienophile and more than the simple Diels-Alder reaction.
To determine the concernedness and asynchronicity of the reaction, the electron in the HOMO of 1,3-cyclohexadiene (diene) is like the outer shell electrons of an atom which can be remove with little energy than any of the other electrons in the molecule.
To rationalize the asynchronicity of the reaction [14] bond breaking as shown in the variation of the change in the bond length in the transition state and the reactants (C3-C5 Δd = +0.043, +0.043 +0.035; and C7-C2 Δd=+0.042, +0.062, +0.060 for H, OH, NO2 respectively) occur first bond making while the (C3-C7 Δd= -0.068, -0.067, -0.064; C2 -C15 Δd=-3.396, -3.416, -2.599 and C5-C18 Δd= -3.395, -3.848, -1.637 for H, OH and NO2 respectively) is lagging behind.
The energy of formation
(Table 7) showed the exothermicity of the reactions was as explained using
Hammonds postulate which states that in exothermic steps it is expected that
the activation complex resembles the adjacent reactants that it is closest in
energy to, as shown in the value of the energy of formation of the transition
state for the Diel-Alder reaction between 1,3-cyclohexadiene and substituted,
unsubstituted ethene is much more closer to that of the reactants than it is to
the products. This is as a result of the formation of the two new ![]() - bonds.
- bonds.
Table 7. Energy of formation (kJ/mol)
|
Elements |
GS |
TS |
PRD |
|
H |
-819312.375 |
-819194.25 |
-819409.50 |
|
OH |
-1016854.125 |
-1016715.0 |
-1016930.25 |
|
NO2 |
-1356329.625 |
-1356235.125 |
-1356429.375 |
Conclusion
Density Functional Theory (DFT) at B3LYP level with 6-311++G (2df, 2P) basis set was used to modelled the influence of electron withdrawing group and electron donating group attached to the dienophile on the kinetics and thermodynamic parameters to show the influence on activation parameters. The calculation indicates that electron withdrawing group (NO2-substituted dienophile) lowers the activation parameters and thereby increasing the rate of the reaction as observed with lower rate constant, k value compared with unsubstituted and OH-substituted dienophile.
References
1. Anslyn E.V., Dougherty D.A., Modern physical organic chemistry, University Science books, 2006 (www.uscibooks.com).
2. Aswany P.N., Suresh A., Vijayakumar G. and Renjith T., Substituent effects on simple Diels-Alder reaction from quantum mechanical calculations, International Journal of Current Research in Chemistry and Pharmaceutical Sciences, 2016, 5 (3), p. 40-47.
3. Balbi N. and Khoumeri B., Thermodynamic and kinetic studies on reversible liquid-phase Diels-Alder reaction between maleic anhydride and 2-methyl furan, Dynamic modelling by thermal analysis, Journal of Thermal Analysis, 1994, 42, p. 461-466.
4. Boger D.L., In HeteroDiene additions: Trost B., M. Ed, Pergamon Press Oxford, 1991, 5, p. 451.
5. Brocksom T.J., Nakamura J., Ferreira M.L., Brocksom U., The Diels-Alder reaction: an update, J. Braz. Chem Soc., 2001, 12, p. 597-622.
6. Celius T.C., Fast Hetero-Diels-Alder reactions using 4-Phenyl-1,2,4-triazoline-3,5-dione (PTAD) as the Dienophile, J. Chem Educ., 2010, 87, p. 10-1.
7. Doering W.V.E., Roth W.R., Breuckman R.F.L., Lennartz, H.W., Fessner W.D. and Prinzbach H., Verbotene Reaktionen - [2 + 2] - Cycloreversion starrer Cyclobutane Chem. Ber., 1988, 121 1-9.
8. Fringuelli F., Tatichi A., Dienes in the Diels-Alder Reactions, Wiley: New York, 1990.
9. Gracia J.I, Martimez-Merino Mayoral J.A. and Salvatella L., Density functional theory study of a Lewis acid catalysed Diels-Alder reaction. The Butadiene + Acrolein Paradgim, J. Am. Chem Soc., 1998, 120, p. 2415-2420.
10. Houk K.N. Gonzalez J., Li Y., Pericyclic reaction transition states: Passions and punctilios, Accounts of Chem. Res. ACS Publications 1995, 28 (81), p. 1935-1995.
11. Juhi M., Tanner D., Recent application of intramolecular Diels-Alder reactions to natural products synthesis, Chem. Soc. Rev., 2009, 28, p. 2983-92.
12. Li, Y., Houk K.N., Diels-Alder dimerizationof 1,3-butadiene: An ab-initio CASSCF study of the concerted and stepwise mechanisms and butadiene-ethylene revisited, J. Am. Chem Soc., 1993, 115 (16), p. 7478-7485.
13. Kersharwani M.K. and Ganguly B., Solvent effect on the stereoselectivity of reaction of methyl acrylate, methyl methacrylate and methyl trans-crotonate with cyclopentadiene: A computational study, Croatica Chemica Acta, 2009, 82 (1), p. 291-298.
14. Norberto K.V.M., Caio F., Teaching thermodynamic, geometric and electronic aspects of Diels-Alder cycloadditions by computational chemistry - An undergraduate experiment, World Journal of Chemistry Education, 2015, 3 (6), p. 141-149.
15. Oppolzer W., In intermolecular Diels-Alder reactions: Trost B., M.Ed, Pergamon Press: Oxford, 1991, 5, p. 315.
16. Jasinski R., Kwiatkowska M., Sharmin V.S., Baranski A., Experimental and theoretical studies of Diels-Alder reaction between methyl (Z)-2-nitro-3-(4-nitrophenyl)-2-propenoate and cyclopentadiene, Monatsh Chem., 2013, 144, p. 327-335.
17. Barone V., Amaud R., Study of prototypical Diels-Alder reaction by a hybrid density functional/Hartree-Fock approach, Chemical Physics Letters, 1995, p. 393-399.
18. Barone V. and Amaud R., Diel-Alder reactions: An assessment of quantum chemical procedures, The Journal of Chemical Physics, 1997, 106, p. 8727-8732.
19. Weinreb S.M., In heterodienophile addition to Diene Trost, B., M.Ed, Pergamon Press: Oxford, 1991, 5, p. 401.